
Today was the last day of my first rotation project in the Davenport Lab at the Wellcome Sanger Institute. While luckily I managed to physically visit the Sanger institute in the first week to meet Emma and get my Sanger laptop just in time as the restrictions started to roll in, the rest of the rotation was working from home. While neither of us knew how this was going to pan out, I think it went really well, and I was definitely in a much more fortunate position than most other PhD students, being able to access all the data I need and perform all analysis remotely. I went in to this rotation not knowing much about sepsis, but now I appreciate the particular challenges researchers are facing in understanding enough about how the dysregulated immune response manifests to be able to accurately diagnose and assist people.
How did I analyse these transcripts?
The main goal of my project was to take the information we had on a large cohort of several hundred patients with sepsis and analyse their messenger RNA transcripts for differential splicing events. Statistically, this means looking for splicing events which are significantly enriched in one group of individuals, but not in another. This knowledge is beneficial as transcripts are the precursors to proteins which perform various functions in our cells, and these differences may give more insight in to how the immune system responds or why the downstream response differ wildly between individuals. To do this I used a program called Leafcutter, firstly to cluster all the splicing events in to groups which overlap any of their genetic material. Then, I computed the extent to which these introns are spliced differently between the groups of people I defined. This was calculated between groups of people defined by their SRS endotypes, the type of infection they had (bacterial and viral), and the gram status for bacterial infections. From here, I compared the proportion of splicing events at each cluster in each individual with their genotypes, to see if there were any associations (SpliceQTLs) using FastQTL.
What did I discover?
Having not previously analysed splicing of transcripts, I wasn’t quite sure what I find. There were many more significant splicing events between the groups compared than I expected, likely due to the large number of samples we had available, which allowed us to pick up even those with small effect sizes. This reinforced the importance of appropriate filtering based on what we cared about, for example this could be all the events to try and infer regulatory networks, or just those with a large biological effect. I opted for the later due to being relatively short on time, but both are interesting questions. It was also notable that only a very small proportion of the genes which were differentially spliced between SRS endotypes (~5%) were also differentially expressed (their abundance in the cell) between these groupings. This shows that splicing events can (and to a large extent do!) occur in genes not differentially expressed between the groups tested, and such changes would not have been detected by earlier methods such as microarrays. In regards to the underlying biology, I found many examples of genes being spliced differently which also had known roles in the control of immunity and inflammation (but no previously record of their differentially spliced variants!). Combining this with the genotype data allowed us to see very clear examples of splicing events being associated with nucleotide polymorphisms in the patient’s genome, but the downstream biological implications of these would need further experiments to validate. Observations of the influence of genetics on splicing also proposed that you can decrease unequal grouping effects of genotypes at particular loci by selecting for splicing events with SpliceQTLs that have balanced genotypes at these loci. Overall, these results show that there are lots interesting differences in the splicing between medically significant groups of individuals, and reinforce the value of transcript-level information. This has also got me thinking about the interplay between genotype and the environment, how this could impact immune regulation and control through the transcriptome. Additionally, the extent to which splicing is controlled by the genome (and how this is semantically encoded), compared to the role of the present environment.
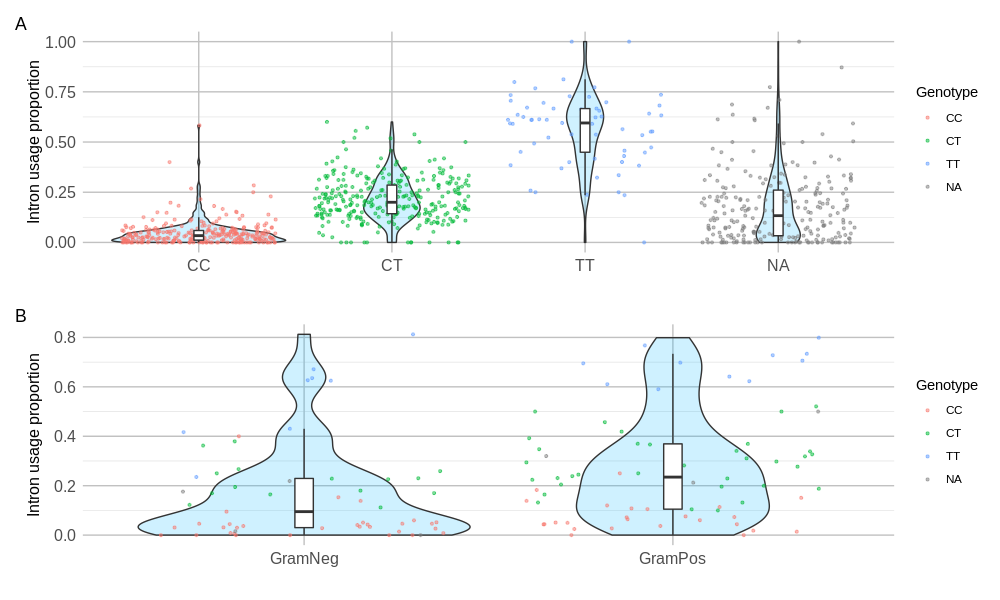
A differential splicing event in the TDRD9 gene. A) Relationship of the most highly significant SNP with associated TDRD9 intron. B) Division and distribution of these same intron usage proportions by the bacterial infection type of the individual.